Interleukin-17 governs hypoxic adaptation of injured epithelium
July 08, 2022
Piotr Konieczny, Yue Xing, Ikjot Sidhu, Ipsita Subudhi, Kody P. Mansfield, Brandon Hsieh, Douglas E. Biancur, Samantha B. Larsen, Michael Cammer, Dongqing Li, Ning Xu Landén, Cynthia Loomis, Adriana Heguy, Anastasia N. Tikhonova, Aristotelis Tsirigos, and Shruti Naik
Science Journal
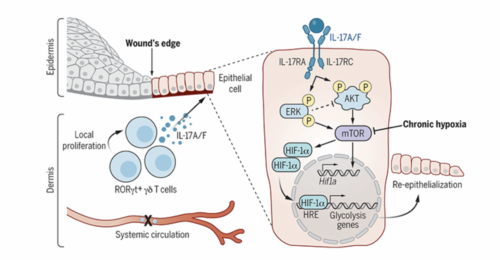
Mammalian cells autonomously activate hypoxia-inducible transcription factors (HIFs) to ensure survival in low-oxygen environments. We report here that injury-induced hypoxia is insufficient to trigger HIF1α in damaged epithelium. Instead, multimodal single-cell and spatial transcriptomics analyses and functional studies reveal that retinoic acid-related orphan receptor γt+ (RORγt+) γδ T cell-derived interleukin-17A (IL-17A) is necessary and sufficient to activate HIF1α. Protein kinase B (AKT) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling proximal of IL-17 receptor C (IL-17RC) activates mammalian target of rapamycin (mTOR) and consequently HIF1α. The IL-17A-HIF1α axis drives glycolysis in wound front epithelia. Epithelial-specific loss of IL-17RC, HIF1α, or blockade of glycolysis derails repair. Our findings underscore the coupling of inflammatory, metabolic, and migratory programs to expedite epithelial healing and illuminate the immune cell-derived inputs in cellular adaptation to hypoxic stress during repair.